
In un famoso disegno Nicolaas Hartsoeker (1656-1725), uno dei primi microscopisti, ritrasse alla fine del XVII secolo una presunta persona in miniatura che egli credette scorgere raggomitolata nella testa di uno spermatozoo osservato al microscopio. È comprensibile che si voglia vedere nel seme l’immagine dell’uomo che da esso trarrà origine.
Il sorgere di una generazione umana dalla precedente avviene attraverso un esilissimo substrato materiale, una coppia di cellule maturate lungo la linea germinale – quantitativamente un nulla rispetto alla mole degli organismi adulti di cui esse fanno parte. E questo si verifica per innumerevoli forme viventi, ognuna infallibilmente secondo la sua specie. Deve esserci, contenuto in un volume così minuscolo, un modello che informerà la costruzione dell’organismo: è comprensibile la fortuna di questa idea, che provocatoriamente potremmo chiamare il mito della rappresentazione interna. Esso, dentro e fuori la scienza, tiene in scacco il nostro sguardo sui viventi.
La sua enunciazione contemporanea risuona in queste recenti parole di Sydney Brenner (1927-…), premio Nobel nel 2002 e uno dei padri della genetica molecolare: «ciò che è passato da un organismo alla sua progenie non è l’organismo stesso, ma una descrizione di esso scritta nel linguaggio molecolare del DNA e dei suoi geni. Tutte le proprietà dell’organismo sono “ridotte” a questa descrizione molecolare» [1].
Il genoma: una banca dati
Ma è proprio vero questo: che nel DNA sono descritte tutte le proprietà di un organismo? che il genoma di ogni organismo, una copia del quale è presente in ogni sua cellula passata e presente, porta scritta una serie di istruzioni in grado di dirigere la sua costruzione e mantenimento? La constatazione di alcuni fatti evidenti ci fa rispondere di no.
Un linfocita, un neurone e un fibroblasto, per nominare solo tre dei circa duecento diversi tipi cellulari nell’uomo, sono entità diversissime fra loro, eppure il loro genoma è identico: che cosa fa la differenza? Difficile immaginare esseri così diversi come una farfalla e il suo stadio larvale noto come bruco, eppure essi contengono, in ognuna delle loro cellule, lo stesso corredo genomico: che cosa fa la differenza, se il genoma è lo stesso?
L’apparente contraddizione sparisce quando il DNA genomico viene considerato per ciò che è in realtà: non un programma, ma una banca dati. Ciò che è scritto nella sequenza del DNA genomico non è una serie completa di istruzioni la cui esecuzione porta all’origine e al mantenimento di un organismo attraverso una azione puntiforme in ognuna delle sue cellule.
In prima approssimazione il genoma, più semplicemente, può essere considerato come composto da pacchetti di istruzioni (geni) ognuno dei quali serve per dirigere la biosintesi di una o più macromolecole di natura ribonucleica (RNA), in un processo denominato trascrizione; a loro volta, alcune di queste molecole di RNA (dette RNA messaggero, o mRNA) possono dirigere la biosintesi ordinata di macromolecole proteiche, in un processo denominato traduzione che si avvale di un sorprendente codice di corrispondenza fra triplette di nucleotidi e singoli amminoacidi (il codice genetico).
Nella sequenza genomica, quindi, sta scritto quali RNA, e quindi quali proteine potranno essere sintetizzate; ma se e in quale misura queste macromolecole verranno sintetizzate nel particolare contesto cellulare in cui il genoma si trova, questo non sta scritto nel genoma stesso. Eppure questo è di fondamentale importanza, poiché l’organizzazione morfologico-strutturale e le caratteristiche funzionali dei diversi tipi cellulari, essenziali per la vita degli organismi pluricellulari, dipendono da quali, delle decine di migliaia di proteine codificate nella sequenza genomica, vengono effettivamente espresse e sono quindi presenti, e a quale concentrazione esse sono presenti (ma non solo da questo, come vedremo).
Una rappresentazione approssimativa ma suggestiva dei diversi modi in cui il genoma viene espresso nei diversi tipi cellulari che compongono un organismo viene fornita da immagini come quelle riportate nella figura che segue, in cui in ogni colonna vengono rappresentati i livelli di espressione individuali (come mRNA) di alcune migliaia di geni sotto forma di barrette rettangolari (una per ogni gene) il cui colore varia da verde (meno espresso) a rosso (più espresso).
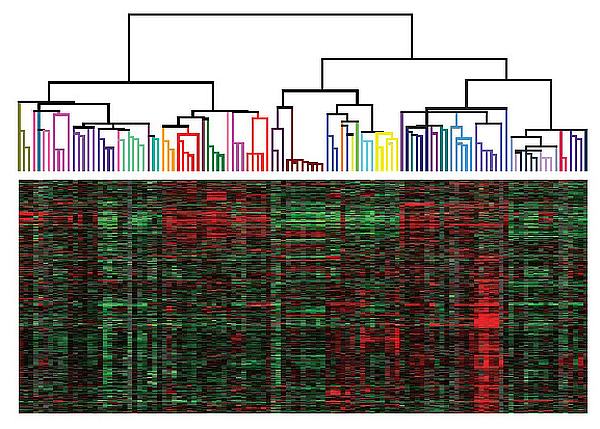 Profili di espressione genica in diversi tipi cellulari umani raggruppati per somiglianza secondo uno schema di «clustering gerarchico»
Profili di espressione genica in diversi tipi cellulari umani raggruppati per somiglianza secondo uno schema di «clustering gerarchico»
Gli scenari della regolazione genica
È facile capire, partendo da immagini come la precedente, che sono innumerevoli i modi in cui la banca dati del genoma può essere utilizzata, un po’ come sono innumerevoli le musiche che possono essere prodotte azionando in modi diversi l’insieme fisso delle canne di un organo.
Se ancora cerchiamo dove stia scritto il «programma», il «progetto» di una cellula e, per estensione, di un organismo, se ci chiediamo dove vada rintracciata una sua eventuale rappresentazione interna, non potremo quindi fermarci al corredo genomico (che assomiglia molto poco al «libro della vita» a cui è stato enfaticamente paragonato), ma dovremo passare ad altre domande.
La prima è naturalmente: da cosa dipende che ognuno dei numerosissimi «pacchetti» di istruzioni (geni) che compongono il genoma venga più o meno (o per niente) espresso? Guidati da questa domanda vediamo schiudersi gli scenari vertiginosi della regolazione genica.
I fattori di trascrizione
L’espressione di un gene dipende innanzitutto dal suo tasso di trascrizione. Ogni gene viene trascritto a opera di un macchinario enzimatico (RNA polimerasi, di cui esistono tre diversi tipi nelle nostre cellule) che agisce sui geni solo nella misura in cui questi sono attivati dal legame, nelle loro vicinanze, di proteine specializzate note come fattori di trascrizione.
I fattori di trascrizione, associandosi in modo specifico e in diverse combinazioni a corti tratti di sequenza di DNA, influenzano positivamente o negativamente il reclutamento o l’attività delle RNA polimerasi sui diversi geni. Nella banca dati del genoma, associata all’istruzione che specifica ogni proteina, c’è anche quella che specifica quali fattori di trascrizione ne controllano l’espressione: il tasso di espressione di ogni gene dipenderà quindi innanzitutto (ma non solo) dalle concentrazioni dei diversi fattori di trascrizione dai cui siti di riconoscimento il gene in questione è circondato.
È interessante notare che nelle cellule umane esistono circa 2000 (!) diversi fattori di trascrizione, e che ognuno di essi, essendo una proteina, viene prodotto grazie all’espressione di un proprio gene, il quale a sua volta è regolato da fattori di trascrizione esattamente come tutti gli altri geni. Esiste quindi una estesa e intricata rete di «regolazione dei regolatori», su cui torneremo più avanti.
Il contesto cromatinico: l’epigenoma
Per capire come agiscono i fattori di trascrizione, è importante tenere presente che il DNA genomico, nelle cellule dell’uomo e di tutti gli altri organismi eucariotici, si trova strettamente associato a un gran numero di macromolecole proteiche a formare un denso aggregato nucleoproteico denominato cromatina.
Nel contesto cromatinico il DNA può essere più o meno condensato e quindi più o meno accessibile. Le più abbondanti proteine della cromatina sono i cosiddetti istoni. Gli istoni H2A, H2B, H3 e H4 si associano a formare un ottamero (contenente due molecole di ogni istone) attorno al quale il DNA si avvolge strettamente a formare con gli istoni il cosiddetto nucleosoma, che rappresenta l’unità strutturale e funzionale fondamentale della cromatina.
Un tratto di DNA facente parte di un nucleosoma tende a essere meno accessibile di un DNA senza nucleosomi, inoltre diversi nucleosomi possono condensarsi in strutture più compatte in cui il DNA è ancora meno accessibile. Una caratteristica intrigante degli istoni è che essi possono essere modificati chimicamente in numerose posizioni (soprattutto mediante acetilazioni e metilazioni reversibili), e alcune di queste modificazioni (o loro combinazioni) possono influenzare localmente la struttura della cromatina, il tasso di trascrizione del DNA stesso, e quindi l’espressione genica.
Se consideriamo il DNA nel contesto cromatinico, esso può apparirci quindi suggestivamente come un genoma, caratterizzato (in modo irreversibile se si eccettua l’insorgenza casuale di mutazioni) da una particolare sequenza di DNA, che serve da supporto alla «scrittura» (stavolta facilmente modificabile) di un altro livello di istruzioni, fatte di profili di modificazioni istoniche o di metilazione in alcuni punti dello stesso DNA, che sono andate confluendo sotto il termine evocativo di «epigenoma» (ciò che sta sopra il genoma, e nello stesso tempo ciò su cui si potrebbe basare una eredità biologica epigenetica, cioè non legata alla trasmissione di una particolare sequenza di DNA).
Ci sono proteine che partecipano alla «scrittura» dell’epigenoma (enzimi chiamati istone acetil-trasferasi e istone deacetilasi, istone metil-trasferasi e demetilasi, DNA metil-trasferasi, eccetera) che introducono o rimuovono modificazioni chimiche degli istoni o del DNA, e proteine alle quali viene attribuita la capacità di «leggere» le istruzioni epigenomiche.
Il risultato di questa lettura può essere l’apertura o chiusura locale della cromatina, o l’alterazione fine della posizione o composizione di singoli nucleosomi, con il risultato importante di una modificata trascrizione dei geni che, per posizione nel genoma, sono interessati da queste alterazioni cromatiniche. Va aggiunto che, generalmente, gli enzimi che scrivono l’epigenoma ne sono anche dei lettori, per cui quello dell’epigenoma è un «testo» la cui lettura ne promuove e condiziona continuamente la riscrittura…e qui ancora una volta la metafora del libro tende a vacillare.
L’epigenoma e lo scenario della vita
Una considerazione molto importante riguardo all’epigenoma è che esso tende a essere diverso e caratteristico quando si passa da un tipo cellulare all’altro, tanto che si è pensato di poter rintracciare nell’epigenoma quella fonte di diversità morfologica e funzionale che caratterizza, per esempio, tipi cellulari diversi che possiedono lo stesso genoma. Addirittura si è fatta strada l’idea che il programma della vita, invano cercato nel genoma, in realtà si trovava lì, appena sopra, nell’epigenoma!
Nella sezione divulgativa di un grosso programma di ricerca finanziato dall’Unione Europea (Epigenome Network of Excellence) si legge che «l’epigenetica conferisce forma alla vita» e a James Watson (1928-…), a cui venne da chiedersi se il DNA, «questa collana di zucchero attorcigliato, contenente perle fatte di basi puriniche e pirimidiniche, non sia in realtà Dio», i moderni epigenomisti sono tentati di rispondere che «l’alfabeto dei geni è come la parola di Dio, e la sua traduzione [ad opera dei fattori epigenetici] è come la sua mano» [2].
È superfluo commentare il grado di confusione che tutto questo può generare, e un po’ inquieti ci si ricorda del monito di Erwin Chargaff (1905-2002), che sul DNA la sapeva molto lunga: «In nessun’altra attività intellettuale la distanza fra ciò che si fa concretamente e le deduzioni che se ne traggono è così grande come nella scienza contemporanea» [3].
Il ruolo determinante dei fattori di trascrizione
Comunque si vogliano enfatizzare le scoperte (pure importanti) sull’epigenoma, anche alla domanda se il programma della vita si trovi nell’epigenoma, la risposta è un deciso no. Ma in questo caso la ragione è molto semplice: i profili epigenomici sono in larghissima misura un riflesso (e non la causa) dei profili di attività dei geni, i quali sono a loro volta determinati dai fattori di trascrizione [4], sui quali quindi conviene che ci soffermiamo ancora un po’. In effetti, fra le conseguenze del legame dei fattori di trascrizione al DNA c’è quasi sempre anche la modificazione dello stato della cromatina, per esempio dovuta al reclutamento, da parte dei fattori di trascrizione, di enzimi che catalizzano acetilazione, deacetilazione, metilazione o demetilazione degli istoni.
Il ruolo centrale dei fattori di trascrizione nella determinazione dello stato di una cellula è stato sottolineato in modo spettacolare dagli esperimenti che hanno fruttato a Shinya Yamanaka (1962-…) il premio Nobel per la Fisiologia/Medicina nel 2012, nei quali fibroblasti (cellule differenziate del tessuto connettivo) di uomo adulto sono stati trasformati in cellule staminali pluripotenti, cioè in cellule diversissime dal punto di vista morfologico e strutturale) semplicemente incrementando in modo artificiale nei fibroblasti la concentrazione di quattro fattori di trascrizione caratteristici delle cellule staminali [5].
La combinazione di questi quattro fattori di trascrizione si è così rivelata in grado di riprogrammare i fibroblasti, e questa riprogrammazione ha comportato come atteso anche quella dei profili epigenomici.
Sulla base di quanto detto finora, a chi ancora sia alla ricerca del programma della vita converrà quindi concentrarsi soprattutto sui fattori di trascrizione, e sulla variazione delle loro concentrazioni nei diversi tipi cellulari, piuttosto che sulle variazioni epigenomiche che ne conseguono. In effetti, si possono riscontrare chiarissimi profili di espressione tessuto-specifica dei fattori di trascrizione, dall’osservazione dei quali si può facilmente concludere che essi sono proprio fra i fattori principali da cui dipende l’espressione genica tessuto-specifica che era così suggestivamente illustrata dalla precedente immagine.
A questo punto un epigenomista di larghe vedute potrebbe osservare che in fin dei conti anche i fattori di trascrizione fanno parte dell’epigenoma. Questo sarebbe – anzi è – uno spunto molto interessante, in quanto ci invita ad alzare lo sguardo e allargare il campo di ciò che è ereditabile da cellula a cellula e da organismo a organismo senza essere DNA o qualcosa di strettamente associato a esso. Come vedremo, una volta disinibito, questo allargamento di campo e di orizzonti dell’ereditabile risulta difficilmente arrestabile.
La regolazione genica post-trascrizionale
L’enfasi data finora – e giustamente – ai fattori di trascrizione non deve però farci dimenticare che la concentrazione di ognuna delle migliaia di diverse proteine (da cui dipendono, come abbiamo ricordato, le caratteristiche morfologiche e funzionali dei diversi tipi cellulari e quindi, in ultima analisi, degli organismi) non dipende solo dal tasso di trascrizione dei corrispondenti geni, ma anche dalla velocità a cui si svolgono numerosi altri processi successivi alla trascrizione e ugualmente richiesti per arrivare alla sintesi della proteina stessa, nonché dal tempo di permanenza di ogni proteina nella sua forma integra prima che si verifichi la sua degradazione.
In altri termini, la concentrazione di ogni proteina cellulare istante per istante è il risultato delle sua velocità di sintesi e di degradazione, e a determinare la velocità di sintesi di ogni proteina ci sono, insieme alle intricate reti trascrizionali di cui si è parlato sopra, ancora più intricate reti di regolazione genica post-trascrizionale.
Ci sono per esempio centinaia di fattori di splicing (proteine ma anche molecole di RNA) che, associandosi al pre-mRNA, condizionano il modo in cui le diverse porzioni codificanti per proteina (esoni) vengono congiunte fra di loro previa eliminazione di porzioni non codificanti (introni), cosicché diverse proteine possono essere sintetizzate a partire dal trascritto primario (pre-mRNA) di un singolo gene; ci sono centinaia di proteine che interagiscono con l’RNA messaggero e ne influenzano positivamente o negativamente la stabilità, altre che ne influenzano la velocità di traduzione da parte del macchinario di sintesi proteica; e sono stati scoperte negli ultimi vent’anni diverse centinaia di minuscole molecole di RNA (denominate microRNA o miRNA) le quali, riconoscendo tratti complementari di sequenza presenti nelle molecole di mRNA, ne possono influenzare la stabilità e la velocità di traduzione. Ma anche questi regolatori (fattori di splicing, di stabilità, di traduzione) sono a loro volta regolati, e possono esserlo a livello trascrizionale (da fattori di trascrizione) o post-trascrizionale (da fattori di splicing, stabilità, traduzione).
Diversi tipi cellulari presentano profili di espressione specifici e distintivi dei fattori di regolazione post-trascrizionale, oltre che dei fattori di trascrizione. Chi cerchi il programma di ciò che anche una sola cellula è, dovrà quindi complicare enormemente il proprio quadro, per integrarvi anche tutti i regolatori post-trascrizionali e le loro relazioni regolative con i fattori di trascrizione. Se a questo punto, limitandoci a un semplice accenno, introduciamo nel quadro anche tutti i diversi fattori proteici (e sono centinaia!) che intervengono nella regolazione della stabilità di altre proteine, tenendo presente il fatto che anche tutti questi regolatori di stabilità possono essere regolati in tutti i modi menzionati, ci viene il sospetto che la ricerca del programma di una cellula sia davvero ardua.
La risposta ai segnali
Arrivati a questo punto, anche se con un po’ di capogiro, possiamo però dirci soddisfatti per avere enumerato e preso in considerazione tutti i fattori che concorrono a determinare la concentrazione di ogni singola specie proteica in una cellula.
Potremmo sentirci rassicurati dall’idea che i fattori in gioco sono moltissimi, ma li teniamo (almeno concettualmente) in pugno. Tuttavia, non abbiamo ancora introdotto un aspetto fondamentale di tutti i sistemi cellulari. Indipendentemente dalla sua concentrazione, l’attività di ogni proteina, e quindi anche l’attività di ognuno delle migliaia di fattori di regolazione genica, può cambiare in risposta a una miriade di segnali provenienti dall’interno o dall’esterno della cellula. Tipicamente, una proteina può essere presente in una cellula, ma in una forma non attiva, e una sua piccola modificazione chimica (per esempio l’aggiunta di un gruppo fosfato) può renderla attiva. Oppure, viceversa, una proteina è attiva e una sua fosforilazione può renderla inattiva senza farne cambiare la concentrazione. E che la proteina venga fosforilata può dipendere da una serie di reazioni intracellulari che ultimamente dipendono dall’interazione di una molecola di segnalazione esterna alla cellula con una proteina recettore presente sulla membrana cellulare. Senza che ci addentriamo nei territori vastissimi della biosegnalazione, possiamo però coglierne una implicazione essenziale: ciò che una cellula è in ogni istante può essere visto come risposta, in quell’istante, a innumerevoli segnali interni ed esterni. I segnali interni sono riconducibili a numerosissimi indicatori dello stato interno di una cellula (concentrazione di molecole di scambio energetico, di ioni, di innumerevoli metaboliti, di molecole proteiche non ripiegate o difettose, presenza di danni al DNA… per menzionarne solo alcuni); i segnali esterni si riferiscono all’insieme ricchissimo delle forme di comunicazione di una cellula con l’ambiente (che, in un organismo pluricellulare, è costituito innanzitutto da altre cellule). Si scivola così sempre di più verso la conclusione sorprendente che il programma di una cellula non è separabile da ciò che la cellula è, nella sua relazione con l’ambiente, cioè con altro da sé. E, per estensione, si è spinti a concludere che il progetto di un organismo non solo tende a coincidere con l’organismo stesso (e quindi è vano parlare di progetto), ma addirittura non si limita a esso, poiché il tentativo di una sua rappresentazione non può prescindere da ciò che è altro dall’organismo stesso (e quindi l’idea di una rappresentazione interna, così convincente in apparenza, rivela la sua insufficienza). Ma allora nell’eredità biologica, da cellula a cellula, da organismo a organismo, che cosa viene trasmesso, se non un piano, un programma? Nel tentativo di una risposta, riscopriamo l’ottocentesco «Omnis cellula e cellula» di Rudolf Virchow (1821-1902), e l’urgenza di una adesione semplice ai fatti che risuona in queste (stranamente) recenti parole del fisiologo Denis Noble (1936-…): «Noi ereditiamo [oltre al DNA] la cellula uovo nella sua interezza. È grazie all’apparato di espressione genica della cellula uovo che il DNA può essere usato per fare altre proteine. È ereditato anche l’insieme completo degli altri elementi cellulari, mitocondri, reticolo endoplasmatico, microtubuli, membrane nucleare e non, e miliardi di specie chimiche in assetto specifico nei diversi compartimenti cellulari» [6]. Una affermazione preziosa, nella sua apparente scontatezza, perché ci invita a cambiare il nostro sguardo abituato a vedere riduttivamente le cellule come una pasta inerte comandata dal DNA. Eppure si tratta ancora di una semplificazione, non fosse altro che per il fatto che, oltre a tutti i componenti chimici e organellari accomunati dall’essere enumerabili e quantificabili, vengono trasmessi nell’eredità biologica anche un ordine, una forma che il nostro sguardo sempre più analitico stenta sempre più a riconoscere come reale.
Vai al sito del Simposio per le slide della Presentazione
Giorgio Dieci
(Dipartimento di Bioscienze, Università degli Studi di Parma)
Indicazioni bibliografiche e sitografiche
Brenner S. (2010). Sequences and consequences. Phil Trans R Soc B 365:207-212
Chargaff, E. (2009). Variazioni su temi della ricerca derivate da frammenti di Pascal e di altri; in Mistero impenetrabile, Lindau, Torino, p. 209.
Ptashne, M. (2013). Epigenetics: core misconcept. Proc Natl Acad Sci USA :7101-7103.
Takahashi et al. (2007). Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell :861-872.
Noble, D. (2008). Claude Bernard, The first system biologist, and the future of physiology. Exp Physiol :16-26.
Leggi anche
- Emmeciquadro-Speciale n°14: Le frontiere e i confini della Scienza/ Il Simposio 2014
- Emmeciquadro-Speciale n°14: Le frontiere e i confini della Scienza/ Allargare la ragione: Galileo e la modernità
- Emmeciquadro-Speciale n°14: Le frontiere e i confini della Scienza/ Il Metodo Scientifico nelle Scienze della Natura. Un'applicazione alla Fisiologia